Upsampling the Signal: Active Learning with Proxy Spaces
September 25, 2025 · Anton Morgunov
TL;DR for ML Researchers
The key technique here is a cheap upsampling method for active learning when your scoring function is prohibitively expensive. Instead of just using the N high-scoring samples you found, you use them as "beacons" in a cheap proxy space (e.g., PCA on molecular descriptors). You then build your next training set by sampling M >> N unevaluated points from the neighborhoods around those beacons. This "neighborhood expansion" makes the feedback loop dramatically more sample-efficient by propagating the sparse, expensive signal across a wider, denser set of training examples.
TL;DR for Chemists
We developed a computational method that iteratively refines a molecular generator to discover compounds for a specific protein target. When aimed at C-Abl kinase, our model not only rapidly increased the population of high-scoring binder candidates but also produced a molecule similar to asciminib without any prior knowledge of it. The method can also be used for targets with no known binders (e.g., Cas9 HNH domain), making it a powerful, general-purpose engine for generation of protein-specific libraries.
TL;DR for the General Public
Searching for a new drug is like trying to find a specific grain of sand on a vast beach. Looking one grain at a time is impossible. Our approach is a new kind of metal detector. It first takes a few random readings, and instead of just beeping, it builds a map of the entire beach, predicting which areas are most likely to contain what you're looking for. It then focuses its search only on those high-probability areas. This makes the process of discovering new potential medicines thousands of times faster and more efficient.
Primary Contributions
Landscape & The Gap
Let's say you're trying to find a molecule that can bind a certain protein. Currently, you:
- start by taking a big, generic library of drug-like molecules
- you pass them through some empirical heuristics- or rule-based filters
- you do some automated assays in the lab
and then maybe, maybe, you have 0.01% of something that could be a hit. This is, obviously, pathetic, so can we do any better? Well, you might think, wouldn't it be good if instead of using a generic, undifferentiated library of molecules that could bind to any of the 100k protein targetshow many proteins are in a human body? nobody knows, really. there are >20k genes, which can be spliced in different ways (accepted number is >100k protein isoforms), which then can have all sorts of post-translational modifications possibly resulting in millions of proteoforms., we could use some library that is somehow specific to our protein?
well, in this day and age, the solution is, obviously, a transformer, but if you have a large enough dataset to train an ML model, then you, by definition, have the desired library.
Why can't we run AutoDock Vina on the generic library?
Good idea, except it'd take at best minutes, if not hours. If you have 5M molecules in your generic libraryyou scramble everything you can find MOSES , ChemBL , BindingDB , and GuacaMol, and assuming average docking takes 30 min, it'd take you 285 years of CPU time. And even if you manage to paralellize it over 1k cores (and not lose sanity), that would be 100 days of wall time.
What about the ML docking solutions?
Ah, the classical let the GPUs go brrr. We looked at DiffDockv1, which was SOTA at the time, which finds a pose from a PDB structure and a SMILES string in just a minute (on an A100). Evaluating whole 5M collection is still not an option (9.5 years on 1 GPU, 30 days on 100 GPUs). Fine, let's train a generic transformer-based molecular generator on these 5M molecules, generate some subset, dock it, and fine-tunefurther train with a smaller learning rate on the high-scorers.
To make sure the model gets some meaningful learning signal and not just memorizes the high-scorers verbatim, you need at least 5-10k high-scoring molecules in the active learning (AL) training set. Let's say the fraction of molecules generated by the undifferentiated model that pass the bar is 20%, that means you have to evaluate at least 25k molecules. Still, that would take 2 weeks on a single GPU.
The Key Insight
For a moment, let's assume we're willing to dock 10k molecules. We have two options:
- Option A: generate 10k molecules and dock them all.
- Option B: generate 100k molecules, sample 10k, and dock that subset.
A somewhat peculiar aspect of generating molecules using stochastic models is that Option B always results in a more diverse set of candidatesa model is sampling from a complex, learned probability distribution, p(molecule). this distribution has high-probability "modes" (common, easy-to-form structures it saw a lot during training) and very low-probability "tails" (rare, novel, or more complex structures). ensuring tails are well represented is a variation of Coupon's Collector problem ..
One of the decisions you now have to make is how do you sample the generated molecules. Just randomly? You'd likely under-represent the rare and potentially more relevant molecules. An alternative strategy would be to create some chemical space proxy and ensure the subset covers all of it. You could do farthest point sampling or you could partition the generated molecules into, say, 100 clusterswe use K-Means clustering with k-means++ as seeding algorithm and then sample from each of those clusters.
In our case, we constructed the chemical space proxyam being pedantic and adding the word proxy to distinguish it from the single, real Chemical Space™ that contains all molecules by projecting RDKit Descriptors (196 in total) into a 120-dimensional space (which preserves >99% of the variance). The descriptors are different physicochemical properties and counters of all sorts of functional groups.
Insight
The key insight is that these physicochemical properties and functional group counters in some way contribute to whether a molecule binds to a certain protein or not. As a result, molecules physically close in this chemical space proxy are likely to have similar binding scoresthis is not always true, in fact, it's a named phenomenon ( activity cliffs ), but we made a bet, and it worked in our case. Maybe because our choice of the binding score is an approximate metric. Or because we had a very diverse set of descriptors.
In this work, we implement and demonstrate the effectiveness of the following active learning strategy:
- Generate 100k molecules and partition them into 100 clusters
- Sample 10 molecules from each cluster
- Dock resulting 1000 molecules with DiffDock (16 hours)
- Calculate a weighted sum of interactions in the binding pose (let's agree to call this a binding score)
- Calculate cluster scores by taking an average of binding scores of molecules from this cluster (avg of 10)
- Construct Active Learning training set by taking high scoring molecules AND sampling 5k extra molecules from clusters that have high scores
- Fine-tune the molecular generator
- repeat steps 1-7
Evidence and Impact
Exhibit A. Evolution of the distribution of generated molecules after 5 iterations of active learning (only 5k molecules are scored, total wall time for the pipeline is 5 days):
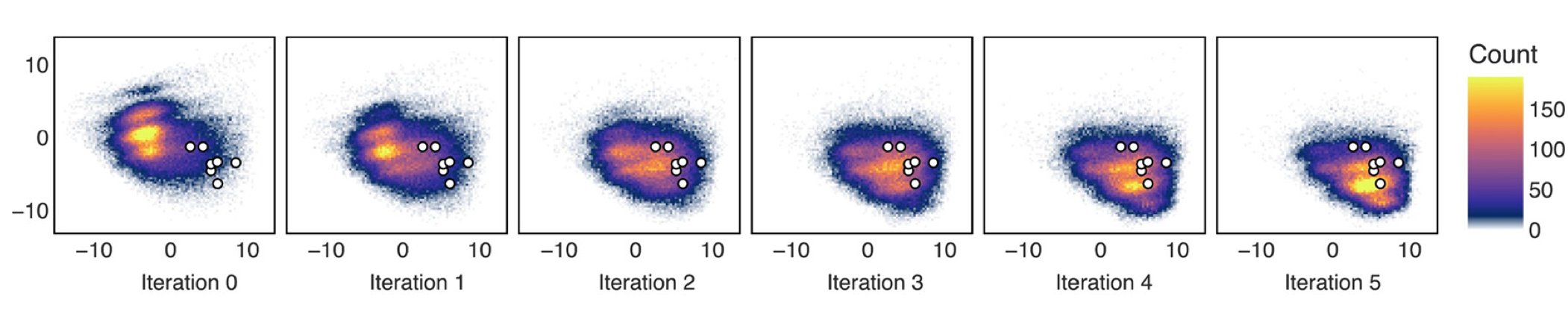
The figure shows the location of generated molecules in the chemical space proxy (only first two principal components are visualized). The white circled dots are FDA-approved inhibitors of C-Abl kinase. As you can see, most of the molecules generated by the baseline model (trained on generic library) are located far away from these inhibitors. And in just 5 rounds of active learning, the distribution shifts dramatically.
It should be noted - we never emphasizedwhat I mean is we didn't enforce sampling or docking or AL training set to include these structures the structures of approved inhibitors as correct solutions. The asciminib is even completely absent from the training setwe chose C-Abl as extra validation target during peer-review, so unfortunately it was too late to remove all FDA targets from the training set, and yet the model learns to generate something very, very close to it!
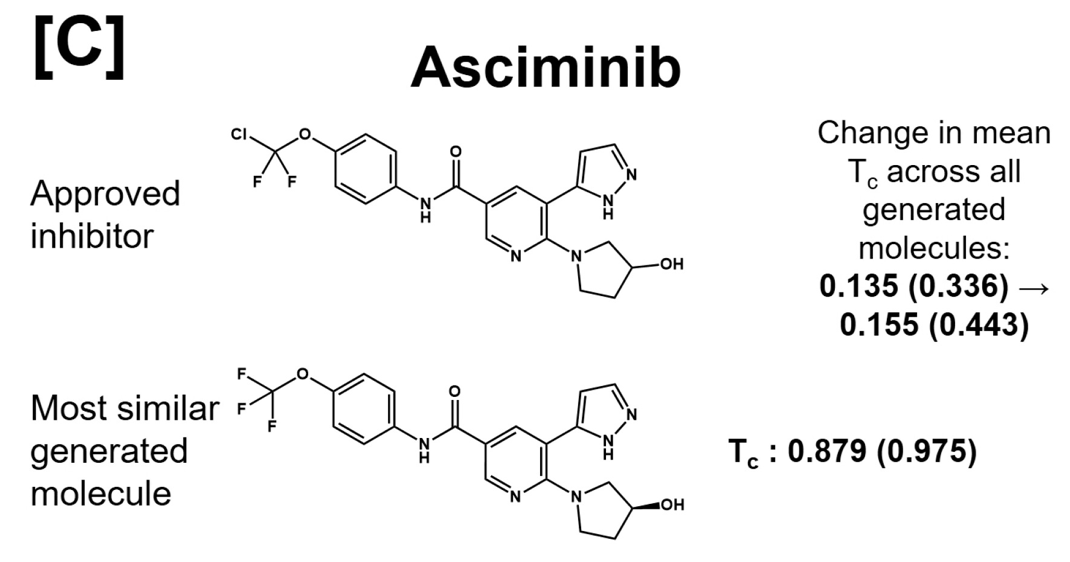
the only difference is that Asciminib contains a OCFCl group, while our model proposes a OCFit could very well be that Novartis landed on OCF Cl after trying to optimize the OCF variant for say biological half-life. meaning, for an initial suggestion, it's a good candidate!
Extending to targets without known binders
For targets without known binders we need a new success criteria, we can't just look at distance to the known binders. So let's dive into technical details and look at our definition of the binding score.
Definition
Binding Score — a top binding pose suggested by DiffDock is passed through ProLIF fingerprinting utility which determines how many ionic interactions, H-bonds, or hydrophobic interactionsthere are a few more interactions reported by ProLIF, see our code for details. are present between protein and a molecule. The binding score is calculated as a sum of these counts with custom weights (7 for an ionic interaction, 3.5 for H-bond, 1 for VdW contactthese are roughly proportional to average free energies of such interactions)
The predictive ability of this binding score is modest - the correlation between this score and the pKd (for targets in PDBBind) is 0.32. However, 99.6% of the protein-ligand pairs in PDBBind have a score above 11, and only 20% of the molecules generated by a baseline model score above 11. In other words, it's not necessarily true that the higher the score, the better the binder, but if your molecule scores below 11, it has very low chance of being a binder at all. So, for the Cas9 HNH runs, we treated all molecules scoring above 11 as high-scoring.

The figure above shows the evolution of the distribution of scores of 1000 molecules after each round of active learning.
| method | number of dockings | training data (source & size) | performance (%>11) |
|---|---|---|---|
| [A] naïve active learning | 5,000 | replicas of ~300 hits | 26% 44% |
| [B] uniform sampling | 5,000 | 300+5,000 sampled uniformly | 28% 51% |
| [C] ChemSpaceAL | 5,000 | 300+5,000 sampled from promising regions | 28% 76% |
[B] is a control, in which the 1k molecules to be scored are sampled from clusters, but instead of sampling 5k extra molecules from high-scoring clusters, you sample 50 from each cluster uniformly. a more interesting control, in my opinion, would be to remove sampling from clusters altogether, but what do I know and Reviewer 2 insisted on this control.
As you can see from the numbers (and visually), our pipeline significantly improves upon naïve AL. The paper contains quite a few extra details and nuances:
- do you just sample from the cluster with the highest score? or from several clusters proportionately to their scores? we chose the latter, but there are still several ways to convert cluster scores to sampling probabilities. We try 3 options: linear, softdiv, and softsub (classical softmax)softmax usually results in most molecules being sampled from 1 cluster, linear doesn't have a clear priority, and softdiv is something in between. Figure S21.1 in SI shows the difference between methods.
- how much do the individual interaction counts change during Active Learning (Figures S21.8-S21.12)
- how much does the model generation capabilities (validity, novelty, uniqueness) degrade over AL cycles (Figure S23.1)spoiler: validity keeps at 99%, uniqueness drops to 80% (you still can generate 100k unique molecules, you just might have to keep trying 125k+ times), and novelty actually increases
Beyond the Paper: Meta-Lessons
This was my first ML paper and the first experience in building software for a non-trivial pipeline, so I made all the mistakes typical for a junior dev.
Jupyter Notebooks are evil
I'm almost ashamedactually, it's also kinda impressive to write this, but the whole project was implemented in a Google Colab notebook. Yeah, I know. Needless to say, I've stopped using jupyter notebooks for anything but quick experiments or data exploration.
For those who haven't gone through the same painful love-and-hate relationship with jupyter notebooks, here is a speedrun:
Defined once, exists forever
let's say you define some variable smile in a cell, try some things, then delete that cell. Until you restart the whole kernel, that variable will still exist in memory. and so when you create a new cell 30 min later and write:
for smiles in smiles_list:
do_something()
another_something()
and_then_something(smile)if you're tired enough, you might not notice that you made a mistake: last line should refer to smiles not smile. With a regular script, you'd get a runtime error. In a notebook however, the code will use the existing definition of smile from half an hour ago. No matter how dumb you think such mistake is and no mater how diligent you'll try to be, eventually you'll go through this process.
They're slow
running the code in jupyter notebooks could be 2-3x slower than from the shellin part as a direct consequence of all variables staying in memory and your RAM being cluttered with junk.
It's just a bad dev experience
At some point you'll have too much code. Yes, you can hide cell content under spoiler, but good luck developing complex pipelines like this without search or click-to-go to function deflike any normal IDE would allow you to. And also you have to worry about scope, because if you define some variableyou could see how this could backfire with some generic names like
molecules
or
smiles_list
that could be used at different stages of the pipeline in Cell 3, that variable will be available in Cell 5, 10, and 20.
It's a bad user experience
I mean, you can just look at the procedure for starting next iteration of Active Learning (from README for ChemSpaceAL):
At this point, you have to go back to Cell 5, change
al_iteration=0toal_iteration=1. Then:
- Run Cells 6, 9, 11, 15, 19.
- Run Cell 21. The AL set loaded should have suffix
al0, i.e. 1 smaller than what's now set in Cell 5 asal_iteration=1.- Run Cell 22. As a sanity check, make sure that the weights loaded will be from previous iteration, and saved to the currently set iteration.
- Run Cell 23.
despite looking like madness, for a pipeline in a jupyter notebook, it's actually a decentin my, not so humble, opinion solution. In my prime, I could launch next iteration for 4 variants of the pipeline running in parallel in just 30 minutes. I could do that because the notebook was optimized to minimize the amount of manual interventions a user had to make. For example: to prepare active learning training set for iteration , you need to provide a path to the file with generated molecules, a file with clusterings, and a file with scored molecules (and the scores). You have two UX choices:
- Ask the user to manually specify those paths. After rounds of active learning, you'd have copies of such files (corresponding to each iteration), so a user would be expected to not mess them uprunning several pipelines (e.g. for different targets) in parallel would be even more challenging.
- Create formulaic names for those files that depend on key dynamic variables and the AL round.
Option 2 seemed really preferable. And had we done everything as a proper python package that you execute through some scripts with some yaml config file, it'd have been perfect. However, because it was a jupyter notebook (in a Colab), you have to manually run all the "config" cellsthe path to scored files can only be constructed from the config for scoring step, which depends on the config for sampling step, which depends on the config for generation step (step 1 from instruction above) that set up all the paths.
So, once you get familiar with how the notebook works, the process is quite efficient and you only need a few Colab credits. But the learning curve is quite steep.
Insight
The real lesson here is that you should learn to build your research tools like production-grade software. Just because you're a chemist (or other non-CS scientist) doesn't mean you're absolved from reading CLRS or 6.031 notes. And not just because that's the right way, but simply because it'll make your life so much easier in the long run.
Tests are not optional
I knew about the paradigm of test-driven development. I knew that unittest or pytest exist. I just didn't have a habit of writing testsI started taking CS classess too late to subject myself to
6.031, and maybe some part of me thought it only mattered if you're a real software engineer, not just some chemist writing scripts. This attitude backfired spectacularly. One notable example: at some point ProLIF changed the api for the key method, and we weren't careful enough to pin a specific version, and the error was handled silently, all ligands received identical scores. We thought something was wrong with fine-tuning or generation (because the changed happened after 4 perfectly working rounds of AL), so we spent quite some time debugging those parts before we looked at the scoring component. Had we had tests for all stages, we would have known where to look.
The importance of research taste
The Supplementary Information for our paper has 60 pages, most of which are filled with different figures. Why so many? Well, this is a complex pipeline, so you have to make so many choices:
- what descriptors do you use? everything from RDKit? Why not add MQN descriptors?a question asked by one of the reviewers
- what value of do you choose for clustering?
- the k-means++ is a stochastic seeding algorithm, meaning you can get slightly different partitions each time. do you run it once? or times? what is ?
- how do you convert the cluster scores into sampling probabilities? linearly? with softmax?
for some reason, I felt obligatory to investigate the impact of every moving part. But doing full scale optimization, one variable at a time, is simply prohibitively expensive. We ended up trying two choices of (10 and 100), running K-Means 100 times (and picking clustering with the smallest variance of cluster sizes), and trying 3 options for conversion of cluster scores into sampling probabilities. In hindsight, that was still too much. If I were to do it again, I'd just be much more brave to make a few subjective decisions and live with them. It does increase the risk (what if you make a poor choice), but the counterfactual would be drowning in the noise of optimization of tiny components that probably don't matter that much.
The ability to make correct choices from first principles and based on intuition is what I'd call research taste. And you can only develop the intuition with experience of starting and completing projects. Point being, you'll probably make quite a lot of unnecessary moves in your research career, but as long as you keep track of which decisions paid off and which didn't, you'll probably get better.
Access & Citation
that's all for today, hope i made you curious enough to check out the paper (or at least the figures).
Cite As
@article{chemspaceal,
author = {Anton Morgunov and Kyro, Gregory W. and Brent, Rafael I. and Batista, Victor S.},
title = {ChemSpaceAL: An Efficient Active Learning Methodology Applied to Protein-Specific Molecular Generation},
journal = {Journal of Chemical Information and Modeling},
volume = {64},
number = {3},
pages = {653-665},
year = {2024},
month = {02},
doi = {10.1021/acs.jcim.3c01456},
addendum = {*A.M., G.W.K. and R.I.B. contributed equally. \href{https://github.com/batistagroup/ChemSpaceAL}{GitHub}}
}